The European Medical Device Regulation (EU MDR) entered into force on 25th May 2017. Together with the 2017 EU In-vitro Diagnostic Device Regulation (IVDR), the MDR replaced the existing directives on 26th May 2021 and on 26th May 2022, respectively. The MDR aims to ensure a high level of safety and health protection for patients, users and third parties, as well as to promote the free movement of medical devices in the market.
According to the changing directives, the requirements for the certification of medical devices in the European healthcare system will follow stricter and uniform rules in the future.
This has implications for all manufacturers of medical devices who are seeking certification within the EU for the first time or whose products have already been successfully established on the market.
On our website, manufacturers and other stakeholders in the medical device sector can find help and extensive information on the changes by the MDR. For more background information on the IVDR or Digital Health Applications (DiGA), please follow the corresponding links.
To ease your transition to MDR or IVDR, the Medical Device Innovation Center (MIC) in Mainz offers you comprehensive support in all aspects – from classifying your products to adapting technical documentation and performing conformity assessments. We work closely with you and our partners to find an individual solution for your specific needs.
Contact us today at info@mic-mainz.de or +49 6131 17-9646 and get comprehensive advice from our experts.
What is the MDR?
Until 26th May 2021, two different directives applied to medical devices in the EU:
- The Medical Device Directive 93/42/EWG (MDD)
- The Active Implantable Medical Device Directive 90/385/EWG (AIMDD)
To unify and simplify the regulatory framework for medical devices, the MDD and AIMDD have been replaced by a single regulation: the EU Medical Devices Regulation (Regulation EU 2017/745 MDR). This regulation covers all medical devices except in-vitro diagnostic medical devices, which are covered by a separate regulation: the EU Regulation on In-vitro Diagnostic Medical Devices (Regulation EU 2017/746 IVDR). The EU MDR entered into force on 25th May 2017, and has been applicable since 26th May 2021, while the IVDR has been applicable since 26th May 2022.
The MDR establishes rules for market entrance, market availability and putting into operation of medical devices intended for human use.
This regulation also applies to clinical investigations conducted within the EU involving medical devices.
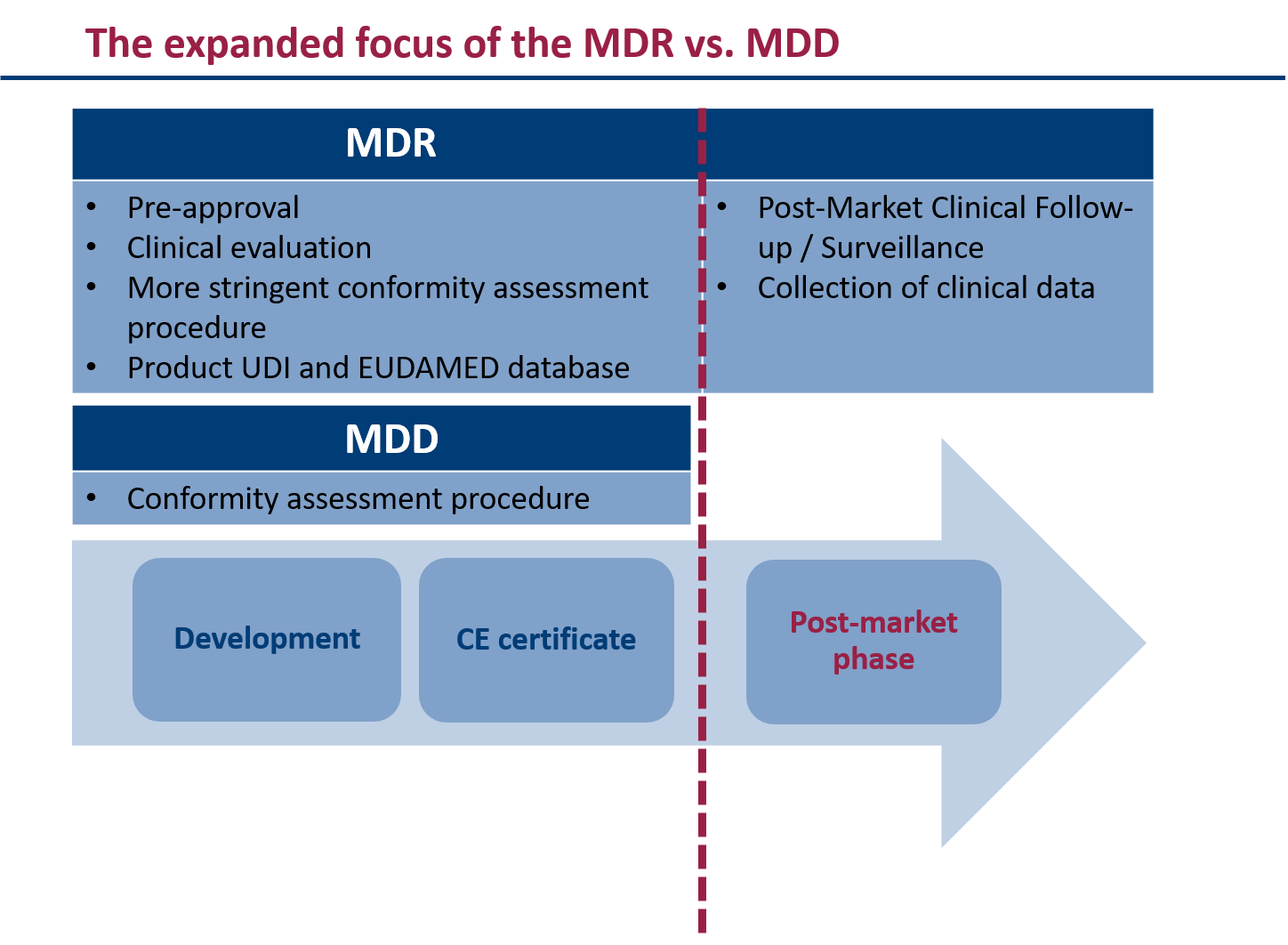
The MDR is more comprehensive and detailed than the MDD and AIMDD. This brings some important changes for manufacturers, notified bodies, authorities and other medical device stakeholders. These include:
- An expanded definition of medical devices that also includes devices without a medical purpose
- A more rigorous conformity assessment procedure with greater involvement of notified bodies
- A system for identifying and tracing devices (UDI – Unique Device Identifier)
- The input of extensive data into the EUDAMED database
- Requirements for the sale of medical devices via the Internet or their distance selling
- Increased requirements for the clinical evaluation and post-market surveillance
- Extended liability rules for importers and distributors
Differences Between the European Directives (MDD/AIMDD) and the Regulation (MDR)
The key distinction between the European Directives (MDD/AIMDD) and the Regulation (MDR) lies in the requirement for member states to transpose the Directives into national law, whereas Regulations have direct and uniform application across all member states, replacing or superseding national laws in the areas where they include provisions that replace or override them. In Germany, national laws govern the implementation of the MDR. In addition, the Medical Device Law Implementation Act (MPDG) supplements the MDR.
(post-market clinical follow-up) in addition to prior authorisation.
The MDR strengthens the requirements for post-market surveillance (PMS) and post-market clinical follow-up (PMCF) of medical devices. Manufacturers must establish a PMS system for each medical device to confirm the safety and performance of the device throughout its lifecycle. In addition, manufacturers will need to collect and evaluate more clinical data to identify side effects and to ensure that the device has an appropriate risk-benefit ratio. Data quality and management must also be optimised.
Transition Periods from the European Directives (MDD/AIMDD) to the Regulation (MDR)
New products are subject to the full scope of the MDR from the start of the certification process. There are different transition periods for already existing products. These may depend, among other things, on the classification of the risk class according to MDD/MDR. According to the proposal to amend the MDR and IVDR adopted by the European Commission on 6th January 2023, the following transition periods now apply:
- The transition period for medical devices that received a certificate or declaration of conformity before 26th May 2021, will be extended by one year. This means that the transition period now applies until 26th May 2025, or until the certificate/declaration of conformity expires.
- The transition period for Class III custom implantable devices and moderate and lower risk devices remain unchanged at 26th May 2026 and 31st December 2028, respectively.
- The sell-off deadlines for medical devices with a certificate or declaration of conformity before 26th May 2021, will be eliminated, meaning these products can be sold as long as they are in stock.
These changes intend to give the manufacturers more time and flexibility to adapt to the new requirements of the MDR and ensure supply. The European Parliament and the Council adopted the proposal on 16th February 2023.
In the following decision tree, you can understand the applicable transition periods for your medical device.
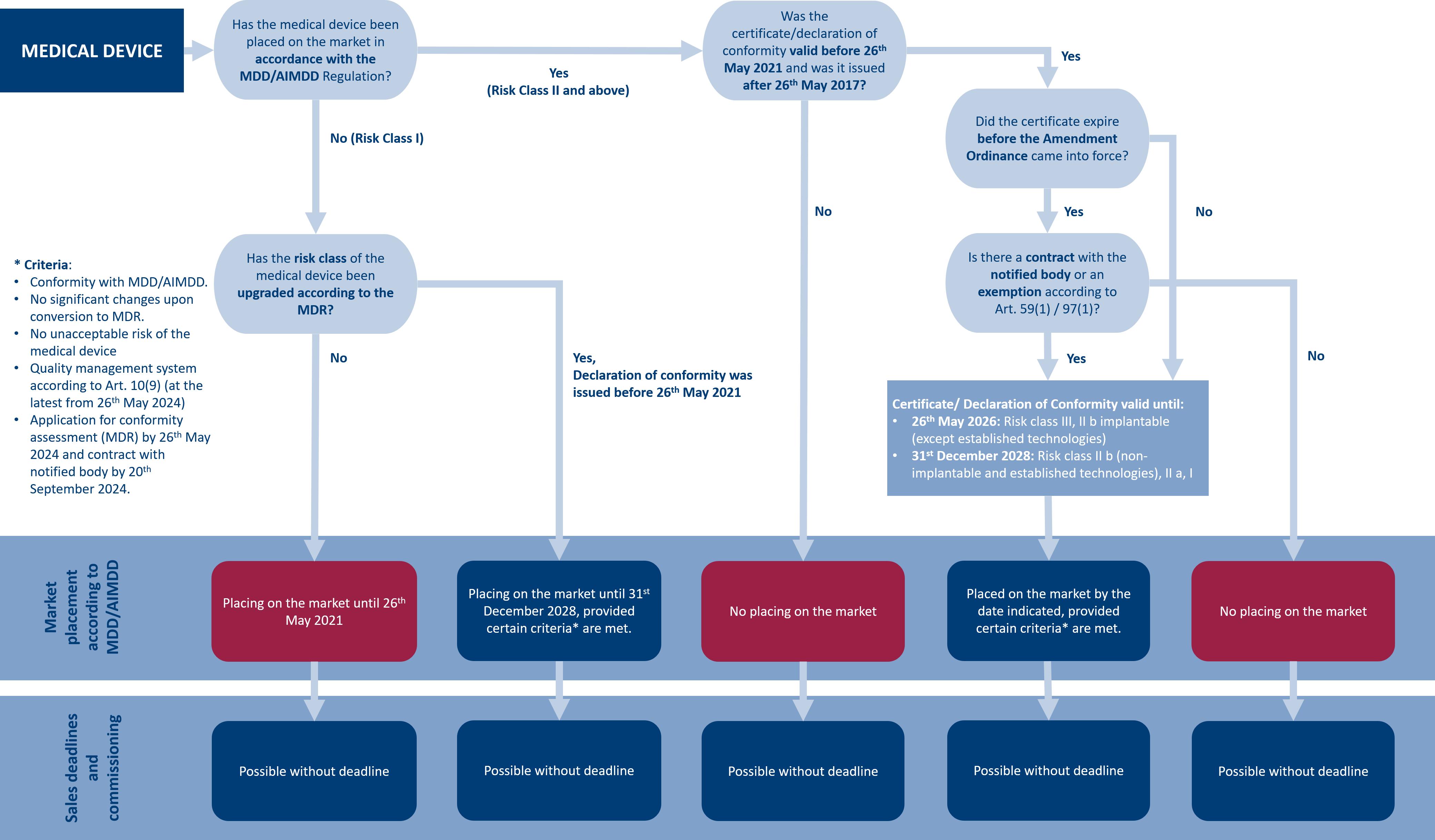
The MDR also requires all manufacturers to implement post-market surveillance (PMS) and post-market clinical follow-up (PMCF) of their medical devices. The processes are mandatory for all medical devices as of the MDR’s effective date and have no transition periods. However, the use of the EUDAMED database remains voluntary and therefore has no transition period.
Other relevant documents for determining medical device transition periods can be found here:
- FAQ on transition periods provided by the working group NAKI (National Working Group for the Implementation of MDR and IVDR) and launched by the German Federal Ministry of Health (BMG).
- FAQ from the CAMD Transition Sub Group (CAMD: „Competent Authorities for Medical Devices“.
- Document 2019-10 from the MDCG (Medical Device Coordination Group). This document sets out considerations for the transition with regard to market surveillance. According to this document, it is possible for a manufacturer to benefit from the transition periods even if its Notified Body has not yet been designated for the MDR. However, the prerequisite is that the Notified Body continues to actively perform market surveillance and act in case of problems.
Requirements for Medical Devices According to EU MDR
The MDR contains many new requirements for medical device manufacturers to ensure the quality and safety of devices for users. Among the most important are the following:
- More and better safety and performance requirements, as well as a higher volume of accompanying information for patients
- Detailed and up-to-date technical documentation
- Regular post-market surveillance of the medical device through post-market surveillance
- A necessary clinical evaluation and testing with clear requirements for the clinical data collected
- Special requirements for medical devices containing hazardous substances
- Unique identification of each device by means of a product identification number (UDI)
Technical Documentation
An important part of the documents that needs to be submitted is the technical documentation of the medical device. It must be clear, understandable and well structured. The technical documentation should contain basic safety and performance requirements and is important for the certification of a medical device.
The elements listed here must be included. Further details can be found in Annex II.
Classification, Approval, Placing on the Market
In addition to the requirements for technical documentation, the new EU MDR also has implications for the classification, approval and marketing of medical devices:
- The MDR changes the risk classes of some medical devices. The classification of old and new products must be adapted to these guidelines.
- The conformity assessment procedure from the MDD no longer exists in the new MDR and is replaced by a conformity assessment from the manufacturer, i.e. an approval without a European medical device authority.
- The MDR leads to an EU-wide standardisation of the activities and test certificates of the notified body (MDR certificate). In addition, a scrutiny procedure will be introduced for Notified Bodies. Notified bodies can thus be obliged to report each new application for conformity assessment for a high-risk product to a committee of experts (the Medical Device Coordination Group, MDCG).
Organisational Requirements in the MDR
In addition to various requirements for the medical device itself, the EU MDR also brings with it some new regulations and specifications for the manufacturer and his company. The most important organisational innovations include:
- The documentation retention period increases from 5 to 10 years.
- Quality management system (QM system): The ISO 13485:2016 compliance requirement must be met and is independent of the risk class of a manufactured medical device.
- The MDR (and IVDR) introduce for the first time a responsible person “for regulatory compliance” who assumes the duties of the safety officer. Both manufacturers and their authorised representatives must designate such a person responsible for regulatory compliance (PRRC).
- The EUDAMED database will be enlarged. In addition to government institutions, notified bodies, the public and manufacturers will also be granted access.
- The MDR distinguishes between several economic operators, with specific requirements for manufacturers, distributors, importers and authorised representatives.
Structure of the MDR
Compared to the MDD, the MDR has been restructured in terms of form and content. In the following, we have listed chapters and annexes of the MDR for you and compiled all relevant links to the original texts of the MDR.
MDR Chapters
- Chapter: Scope and definitions
- Chapter: Requirements for manufacturers, distributors and member states: Conformity assessment procedures, labeling, post-market clinical follow-up, post-market surveillance, and more
- Chapter: Traceability of products, esp. UDI
- Chapter: Requirements for Notified Bodies
- Chapter: Classification and Conformity Assessment
- Chapter: Clinical evaluation and clinical investigations
- Chapter: Market surveillance, notification system
- Chapter: Cooperation of Member States, Medical Device Coordination Group and other Experts
- Chapter: Confidentiality, Data Protection, Penalties
- Chapter: Transition periods and more
MDR Annex
- General safety and performance requirements
- Technical documentation
- Technical documentation for post-market surveillance
- EU declaration of conformity
- CE marking of conformity
- Information to be submitted for registration of devices and economic operators (UDI)
- Requirements for notified bodies
- Criteria for classification
- Conformity assessment based on quality management system and assessment of technical documentation
- Conformity assessment on the basis of type examination
- Conformity assessment on the basis of product conformity verification
- Procedure for custom-made products
- Certificates issued by a notified body
- Clinical evaluation and PMCF
- Clinical investigations
- List of product groups without medical purpose
- Correspondence table
Download Area
| EU original versions |
| German (HTML) |
| English (HTML) |
| DEUTSCH (PDF) |
| ENGLISCH (PDF) |
Your Checklist for the Change from MDD to MDR
✔ Get to grips with the contents of the MDR (See DOWNLOAD AREA) and familiarise yourself with the procedures that will be mandatory in the future.
✔ Check your entire product portfolio for the need to update about the new requirements.
✔ Update the Technical Documentation according to the MDR requirements.
✔ Check what sort of clinical data is missing to prove the requirements of the MDR for the individual product groups. If necessary, these can still be collected as part of PMCF studies under the old directive.
✔ Critically review your quality management documentation regarding the new MDR requirements. Consider how to ensure traceability throughout the manufacturing and supply chain.
✔ Create a plan to ensure product PMS. A key point of the MDR is the post-market surveillance of products. This also has implications for product liability.
✔ Since the MDR requires a unique product identifier, you should think about its affixing and assignment at an early stage.
✔ The tasks of the previous safety officer are expanded considerably. The regulation refers to a compliance officer with very broad compliance responsibilities for manufacturers and authorised representatives.
✔ Consider the necessary human resources and qualification requirements for training and relevant professional experience in your time and budget planning.
✔ For more information on the MDR, refer to the Medical Device Coordination Group’s guidance papers published on various topics. These guidance papers are not legally binding, but reflect recommendations at the EU level.
There is a need for immediate action!
Do you need support in this regard or are you interested in individual consulting so that you can successfully implement the MDR specifically for your products?
Contact us today at info@mic-mainz.de or +49 6131 17-9646 and get comprehensive advice from our experts.