Die neue EU-Verordnung für In-vitro-Diagnostika (Verordnung EU 2017/546, IVDR) ist eine Verordnung der Europäischen Union, die die Anforderungen an In-vitro-Diagnostika (IVD) regelt. Sie trat am 26. Mai 2017 in Kraft und ersetzte die frühere EU-Richtlinie über In-vitro-Diagnostika (Richtlinie 98/79/EG, IVDD). Für Hersteller galt eine Übergangsfrist von fünf Jahren, bis zum 26. Mai 2022, um die überarbeiteten IVDR-Anforderungen für CE-gekennzeichnete In-vitro-Diagnostika umzusetzen. Die Verordnung ist in allen Mitgliedsstaaten der Europäischen Union und der Europäischen Freihandelsassoziation (EFTA) rechtskräftig.
Anforderungen der IVDR
Die IVDR stellt höhere Anforderungen an die Hersteller, um die Sicherheit, Leistungsfähigkeit und Nutzen (sog. Leistungsbewertung) der Produkte zu gewährleisten. Die Verordnung sieht u.a. eine strenge Überprüfung der technischen Dokumentation und klinischen Nachweise vor, um sicherzustellen, dass die IVD-Produkte die grundlegenden Sicherheits- und Leistungsanforderungen erfüllen. Die Hersteller müssen auch ein Qualitätsmanagementsystem vorweisen und eine kontinuierliche Risikobewertung durchführen sowie die Sicherheit ihrer Produkte über die gesamte Lebensdauer nachweisen.
Die IVDR bringt auch Änderungen für die Benannten Stellen, die für die Zulassung der IVD-Produkte zuständig sind. Die Benannten Stellen müssen nun höhere Anforderungen erfüllen und werden regelmäßig von den nationalen Behörden überwacht. Die Überwachung der IVD-Produkte wird auch verschärft. So ist ab sofort bei Produkten der Risikoklasse B (mittlerer Risikograd) und höher (Risikoklasse C und D) eine Benannte Stelle einzubinden. Dadurch steigt automatisch die Anzahl der zu überwachenden Produkte.
Die IVDR stellt somit eine große Herausforderung insbesondere für die Hersteller von IVD-Produkten dar. Sie bietet aber auch Vorteile für Patient:innen und das Gesundheitswesen. Die strengeren Anforderungen an die IVD-Produkte werden dazu beitragen, dass nur hochwertige und sichere Produkte auf den Markt gelangen. Dies wiederum gewährleistet, dass Patient:innen eine präzise Diagnose erhalten und die Behandlung entsprechend angepasst werden kann.
Klassifizierung nach der IVDR
Alle In-vitro-Diagnostika werden gemäß IVDR in die Risikoklassen A, B, C oder D eingeordnet, statt bisher in zwei Listen (A und B). Der Risikoklassifizierung folgt definierten Klassifizierungsregeln und richtet sich hauptsächlich nach der Zweckbestimmung der Geräte und den daraus resultierenden zu erwartenden Risiken. Dabei gilt: je höher das individuelle Risiko und je höher das öffentliche Risiko bei Anwendung des Produkts, desto höher ist die Risikoklasse.
Es ist sehr wichtig, die Klasse des Geräts bereits in frühen Entwicklungsstadien zu kennen, denn nach den neuen Vorschriften ändert sich das anwendbare Konformitätsbewertungsverfahren je nach Klasse. Weitere Informationen zu der korrekten Klassifikation von Medizinprodukten und In-vitro-Diagnostika finden Sie hier.
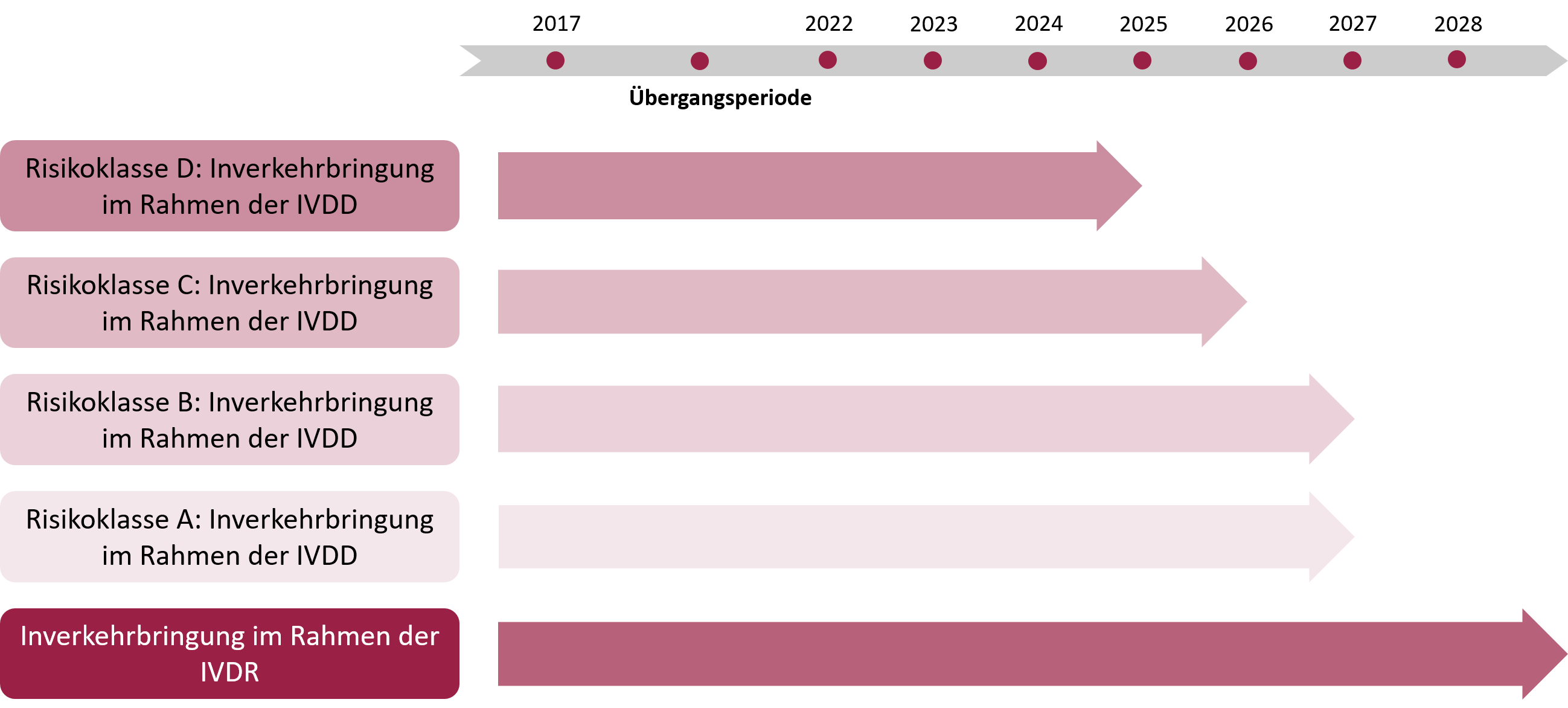
Übergangsplan
Gemäß der Verordnung (EU) 2023/607 des Europäischen Parlaments und des Rates vom 15. März 2023 wurden die Übergangsbestimmungen für In-vitro-Diagnostika angepasst, um Lieferengpässen und potenziellen Versorgungsproblemen im Gesundheitssystem vorzubeugen.
Die Übergangsfristen der Verordnung (EU) 2023/607 ermöglichen es, Produkte, die den Anforderungen der bisherigen In-vitro-Diagnostika-Richtlinie (IVDD) entsprachen, unter bestimmten Bedingungen weiterhin in Verkehr zu bringen oder in Betrieb zu nehmen, während sie schrittweise den Vorgaben der Verordnung (EU) 2017/746 (IVDR) angepasst werden.
Die Übergangsfristen richten sich nach der Klassifizierung gemäß der Verordnung (EU) 2017/746 sowie nach bestimmten Bedingungen, die erfüllt sein müssen:
Produkte, die vor dem 26. Mai 2022 auf den EU-Markt gebracht wurden:
- Produkte ohne Beteiligung einer Benannten Stelle (selbst deklariert):
- 31. Dezember 2027: Ende der Übergangsfrist für Produkte der Klasse D.
- 31. Dezember 2028: Ende der Übergangsfrist für Produkte der Klasse C.
- 31. Dezember 2029: Ende der Übergangsfrist für Produkte der Klasse B und für Produkte der Klasse A, die steril sind.
- Produkte mit Zertifizierung durch eine Benannte Stelle:
- Dürfen bis 31. Dezember 2027 weiterhin in Verkehr gebracht oder verwendet werden, vorausgesetzt, die Bedingungen für die verlängerte Übergangsfrist werden erfüllt.
Bedingungen für die Nutzung der verlängerten Übergangsfrist:
- Bis 26. Mai 2025: Einrichtung eines IVDR-konformen Qualitätsmanagementsystems (QMS).
- Bis 26. Mai 2025, 2026 oder 2027: Antragstellung auf Konformitätsbewertung.
- Bis 26. September 2025, 2026 oder 2027: Abschluss einer schriftlichen Vereinbarung mit einer Benannten Stelle und Übertragung der Überwachung.
- Keine signifikanten Änderungen am Design oder an der Zweckbestimmung.
- Produkte müssen weiterhin den Anforderungen der bisherigen EU-Gesetzgebung (IVDD) entsprechen.
- Konformität mit den Anforderungen der bisherigen Richtlinie 98/79/EG (IVDD).
Quelle: Europäische Union (2023). Verordnung (EU) 2023/607 des Europäischen Parlaments und des Rates vom 15. März 2023 zur Änderung der Verordnungen (EU) 2017/745 und (EU) 2017/746 hinsichtlich der Übergangsbestimmungen für bestimmte Medizinprodukte und In-vitro-Diagnostika. Verfügbar unter: https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=OJ:L_202401860.